
The IO Revolution, Part 2 of 4: The Role of the Host in the Cancer Immunity Cycle
Part one of this four-part series looked at how host, tumor, and environmental factors shape tumor-immune system interactions. In the next few blogs, we will consider how each of these sources of inter-patient heterogeneity has the potential to influence the interaction between cancer and the immune system, the implications of this interaction on immuno-oncology (IO) biomarker and drug development, and—ultimately—the impact on clinical patient management.
An individual’s genetic makeup can greatly influence the odds of developing cancer directly (for instance, germline mutations affecting BRCA1/BRCA2 genes involved in DNA repair) or indirectly (by affecting the immune responses against cancer). The immune system plays a vital role in controlling tumor growth, as evidenced by the increased rate of tumor formation and tumor growth in mice lacking one or more components of the immune system1. Chen and Mellman elegantly describe the interactions between cancer and the immune system as a multi-step, multi-tissue, highly-regulated process, which they named the “Cancer Immunity Cycle” due to the intrinsic self-propagating nature of the process. (Figure 1)19.
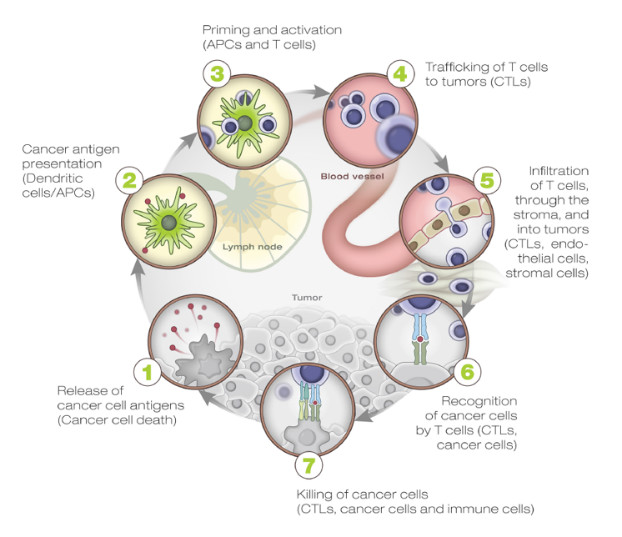
Many factors, both cellular and humoral as well as both stimulatory and inhibitory in nature, are involved in each of the cycle steps. These take place in different tissues, primarily the tumor, the draining lymph node(s), and the peripheral blood. For this reason, any assessment of this process for research and/or clinical purposes needs to comprehensively address this complex biology as a whole instead of looking at single elements of it in isolation.
A brief description of the steps involved in the cancer immunity cycle highlights the main regulatory elements which represent both biomarkers and important targets for therapeutic immune manipulation:
- Step 1 takes place in the tumor where neoantigens, created by the oncogenesis process itself, are released by tumor cells and captured by the professional antigen-presentation cells, i.e., dendritic cells (DCs). The DCs are the master regulators of the antigen-specific immune responses. On one hand, they are required for antigen presentation to naïve T cells, and on the other hand, they carefully “coach” the immune system to respond to “non-self” antigens while maintaining “self” tolerance. The local presence of “immunogenic signals,” such as pro-inflammatory cytokines and factors released by dying cells or by the gut microbiota (acting through Toll-like-Receptor (TLR) agonists), control their level of maturation and antigen-presentation efficiency.
- Steps 2–3 take place in the tumor-draining lymph node(s) where DCs present the captured antigen on major histocompatibility complex I or II (MHCI or MHCII) molecules to T cells, resulting in the priming and activation of effector T cells against cancer-specific antigens. An important regulator of T-cell activation is CTLA-4, a transmembrane protein expressed on the surface of T cells upon activation, which, when bound to its natural ligands (expressed by DCs), results in T cell inhibition, reduced cytokine production, and T-cell proliferation. The checkpoint function of CTLA-4 is primarily of importance within the lymphoid compartment and is mostly limited to the activation of naïve T cells (central tolerance). The importance of this mechanism in modulating immune response to cancer is shown by the clinical success of ipilumumab in metastatic melanoma. The nature of the immune response (immunogenic versus tolerogenic) is determined at this stage.
- Step 4 takes place in the peripheral blood, where activated effector T cells traffic from the lymph node(s) to the tumor. Tumor vascularization and local chemokine gradients are likely to be important factors affecting antigen-specific activated T cells trafficking to the tumor.
- Step 5–7 take place in the tumor microenvironment. Activated effector cells infiltrate the tumor bed following adhesion molecules and chemokine gradients, specifically recognizing and binding to cancer cells through the interaction between its TCR and cognate bound to the MHCI, and kill the target. Killing of the cancer cells releases additional tumor-associated antigens to increase the breadth and depth of the response in subsequent revolutions of the cycle. These last steps in the cycle are controlled by local (peripheral) inhibitory factors, including the PD‑1/PD‑L1 axis (targeted by inhibitory monoclonal antibodies such as nivolumab, pembrolizumab, atezolumab, and darvolumab) as well as TIM-3/LAG-3 receptors, soluble inhibitory molecules such as IDO, etc. The role of these checkpoint molecules is to dampen an ongoing immune response in the periphery with the objective of controlling tissue damage after inflammation. Consequently, the T cells influenced by PD-1 blockage represent a more restricted repertoire than those affected by CTLA-4 blockade. This may explain why immune-adverse events are less frequent when inhibiting the PD-1/PD-L1 pathway than CLTA-4 inhibition.
The genetic variability of the host is an important component of determining the relative success of different cancer epitopes in triggering an immune response, even though it has been less systematically studied.
Generally speaking, this happens at two levels:
- Host variation in specific antigen recognition: MHC alleles are highly polymorphic—different hosts usually have different MHC genotypes and therefore recognize different spectrums of epitopes.
- Genetic polymorphism in immune regulation: Hosts vary genetically in many of the controls of immune response described above. This variation leads to differences in the thresholds that trigger immunity and in the intensity of particular immune effectors deployed against immune targets. Regulatory polymorphisms can cause significant quantitative genetic variability between hosts in the regulation of the immune response, as shown by multiple publications describing the effects of SNPs in immune cytokines in disease prognosis (e.g., follicular lymphoma, prostate cancer, etc.), and/or being associated with response to immunotherapeutic approaches (e.g., Rituxan-Fc-gamma SNPs or IFN-alpha in melanoma patients).
In cancer patients, the cancer immunity cycle does not perform optimally; however, the rate-limiting step or steps in any given patient can be different and multi-factorial. The goal of cancer immunotherapy is to initiate or reiterate a self-sustaining cycle of cancer immunity, enabling it to amplify and propagate an anti-cancer response without unleashing unrestrained autoimmune response. At the level of the single patient, this means selectively identifying and targeting the rate-limiting step or steps.
Tools that enable precise and simultaneous characterization of these relevant analytes are needed because of the multiple levels of regulation that exist to calibrate the nature and magnitude of the molecular changes that govern the immune response (which can be monitored at the level of RNA and proteins) and the hardwire-coded (DNA) individual variations in these regulators.
NanoString Technologies has responded to this need by developing a series of products that enable comprehensive profiling for research of the immune response through the assessment of different analytes (RNA, DNA, proteins) in different tissue types.
In the next Perspectives on Immuno-Oncology blog, we will focus on the tumor-related mechanisms of immune evasion.
REFERENCES
- Kim et al. (2007) Cancer immunoediting from immune surveillance to immune escape Immunology 121(1): 1–14.https://nanostring.com
- Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39(1):1-10.
- Goc et al. (2013) Characteristics of tertiary lymphoid structures in primary cancers. OncoImmunology 2:12, e26836
- Hanahan and Weinberg (2011) Hallmarks of cancer: the next generation. Cell 144(5):646-74.
- Spranger et al. (2015) Melanoma-intrinsic β-catenin signaling prevents anti-tumour immunity. Nature 523(7559):231-5.
- Peng et al. (2015) Loss of PTEN promotes resistance to T cell mediated immunotherapy. Cancer Discovery 6(2):202-16.
- Peng et al. (2015) Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527(7577):249-53.
- Alexandrov et al. (2013) Signatures of mutational processes in human cancer. Nature 500(7463):415-21.
- Dawson MA and Kouzarides T. (2012). Cancer Epigenetics: From Mechanism to Therapy. Cell 150: 12-27.
- Miao M et al. (2015) Molecular Pathways: At the Crossroads of Cancer Epigenetics and Immunotherapy. Clinical Cancer Research 21(18):4040-7.
- Wang et al. (2015) MyD88 Adaptor-Dependent Microbial Sensing by Regulatory T Cells Promotes Mucosal Tolerance and Enforces Commensalism. Immunity 43(2):289-303
- Telesford et al. (2015) A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes 6(4):234-42.
- Sarrayayrouse et al. (2014) CD4CD8αα lymphocytes, a novel human regulatory T cell subset induced by colonic bacteria and deficient in patients with inflammatory bowel disease. PLoS Biology 12(4):e1001833
- Vétizou et al. (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350(6264):1079-84.
- Sivan et al. (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350(6264):1084-9
- Iida et al. (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342(6161):967-70.
- Koti et al. (2015) A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. British Journal of Cancer 112(7):1215-22.
- Sivendran et al. (2014) Dissection of Immune Gene Networks in Primary Melanoma Tumors Critical for Antitumor Surveillance of Patients with Stage II–III Resectable Disease. Journal of Investigative Dermatology 134:2202-11.
- Loi et al. (2015) RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clinical Cancer Research. doi: 10.1158/1078-0432.CCR-15-1125.


